Print Friendly
The Stress Axis Gone Awry: A Possible Neuroendocrine Explanation
for Increased Risk of Completed Suicide
John Michael Bostwick, MD
Dr. Bostwick is associate professor of psychiatry and
director of medical school psychiatry education at the Mayo Clinic College of
Medicine, and a consultant in psychiatry at the Mayo Clinic, both in Rochester, Minnesota.
Disclosure: Dr. Bostwick reports no
affiliation with or financial interest in any commercial organization that
might pose a conflict of interest.
Acknowledgment: Dr. Bostwick appreciates
the help of Christopher Sola, DO, for graphic design; R. Theresa Steele for
secretarial support; and David Mrazek, MD, for ongoing mentorship and
encouragement.
Please direct all correspondence to: John Michael
Bostwick, MD, Mayo Clinic, 200 First St SW, Rochester, MN 55905; Tel: 507-284-2511; Fax: 507-284-4158; E-mail: [email protected].
Focus Points
• Nonsuppression
on the dexamethasone suppression test (DST) may indicate a population with a
much elevated lifetime suicide prevalence.
• Hypothalamic-pituitary-adrenal
axis hyperactivity, as measured by the DST, indicates that a patient may be at
greater risk for more severe forms of depressive illness, including melancholia
or psychotic depression.
• While not
useful as a screening tool or specific diagnostic indicator, a patient’s DST
status provides useful information about the character of the depressive state,
particularly as it relates to agitation and anxiety.
Abstract
While the dexamethasone
suppression test (DST) has not proven useful as a diagnostic test for “true”
depression, it shows increasing promise as a potential indicator of psychiatric
patients at increased risk for suicide. An indicator of the activity level of
the hypothalamic-pituitary-adrenal (HPA) axis, the DST is an endocrine test
that has caught the interest of psychiatric researchers throughout the last
century because of the role of the HPA axis in managing an organism’s
homeostasis and stress response. This article will present a brief literature review
of the role of the DST in psychiatry. It will propose a hypothesis, supported
by data from two recent small studies, that suggests that chronic overdrive in
the HPA axis, as manifested in a nonsuppressing DST, may result in an increased
likelihood of suicide when the individual is acutely stressed.
Introduction
Could depression and even
suicide be construed as neuroendocrine problems? Since the 1960s, abnormalities
in the hypothalamic-pituitary-adrenal (HPA) axis have been associated with
severe depressive states, and neuroendocrine interactions have been a logical
place to look for evidence of biology’s hegemony over psychology.1
As biological psychiatry began to assert its authority over the psychodynamic
school during the 1970s and the 1980s, its “holy grail” became an objective
test to prove depression’s presence or absence. Proponents proposed the
dexamethasone suppression test (DST), a measure of HPA axis activity, as a
clinical diagnostic test to distinguish definitively between biologically driven
melancholic depression and psychologically dominated depressive neurotic
states. According to the dogma, when given a dexamethasone challenge, neurotic
depressives should suppress the HPA axis, while “real” melancholic depressives
should not.
The HPA Axis and
Mental Status
The HPA
axis is crucial to survival. At baseline, it maintains homeostasis and
circadian rhythms through a fine-tuned feedback loop, consisting of the
paraventricular nucleus (PVN) of the hypothalamus, the anterior pituitary, and
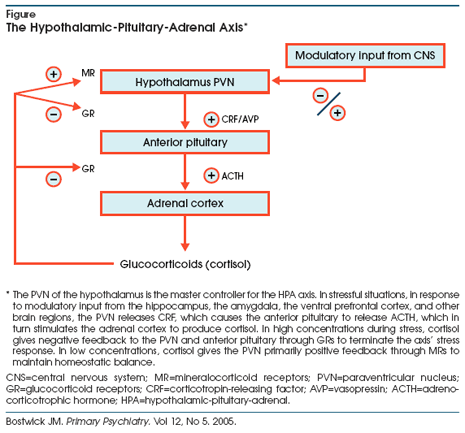
the adrenal cortex. The PVN is the HPA axis master controller, receiving
modifying input from the hippocampus, the prefrontal cortex, and other brain
regions, as well as the axis itself (Figure).
In humans, cortisol is the
hormone messenger that feeds back to the PVN via two receptors. High-potency,
type-I mineralocorticoid receptors that preferentially bind cortisol in
low-concentration environments give predominantly positive feedback at
baseline, fine tuning homeostasis in cardiovascular, metabolic, and immune
systems.2
Under stressful conditions,
however, massive amounts of cortisol are produced. Mineralocorticoid receptors
are saturated, and low-potency glucocorticoid receptors located on the PVN and
anterior pituitary dominate. They offer negative feedback to contain the
all-out stress response. At these times, hyperarousal trumps homeostasis. The
individual demonstrates heightened alertness, decreased libido, insomnia, and
cognitive changes in the throes of a “fight-or-flight” mode.
The HPA axis, the so-called “stress axis,” has long been linked to effects on mental status. Cannon3
observed as early as 1939 that the adrenal glands respond to stress with
increased production, and Cushing4 noticed a link between endocrine
overproduction and “psychic disturbances” in 1913. This stress response is a
normal and necessary protective response in the arsenal of survival weapons.
When hyperarousal and consequent cortisol excess become chronic, however, the
brain is altered permanently, as has been shown in diverse experiments with
rats, macaque monkeys, and humans. The force behind these permanent changes,
both experimentally and naturalistically, is neglect or abuse early in
development. The hippocampus, an inhibitory influence on the hypothalamus,
loses volume and efficacy, and the hypothalamus itself undergoes receptor
changes associated with nonsuppression status and HPA axis hyperarousal.5
Many features of the stress
response are identical to neurovegetative symptoms of depression when chronic.
In the 1960s and 1970s, Sachar,6 Sachar and colleagues,7
Bunney and colleagues,8 and Gibbons9 explored the
relationship between depressive pathology and elevated urine and plasma
corticosteroids. They found particularly strong correlations between
corticosteroid abnormalities and more severe forms of depressive illness,
particularly psychotic depression. The DST, already used to diagnose Cushing’s
disease, opened a window on disturbed HPA axis feedback in severe depression.10
The DST and
Depression
The DST
works in the following way: control subjects who are given a 1 mg dose of the
synthetic steroid dexamethasone at 11pm will typically show subnormal cortisol levels in blood drawn the next day,
indicating appropriate suppression of the HPA axis at the level of the hypothalamus.
HPA axes in a significant proportion of depressed subjects, however, “escape,” and fail to demonstrate a suppressed cortisol level. Thus, the DST was heralded
as a specific laboratory test for the diagnosis of melancholia, a form of
depression with prominent neurovegetative signs.11 Carroll and
colleagues11 asserted in 1981 that “extensive evidence validates
this practical test for the diagnosis of melancholia.”
Writing about the
controversy of articulating between the differential diagnoses of depressive
disorders, Carroll and colleagues11 distinguished between “the
classical condition, melancholia,” also known as endogenous depression, and the
related condition of nonendogenous depression, a term synonymous with
“neurotic, reactive, or characterological” depression.11 “Some
authorities,” they wrote, “insist that there is no demonstrated difference,”
but they asserted that they had proven them wrong with “a test of
neuroendocrine function that is highly specific for melancholia”11 (ie,
the DST).2
Another article,12
typical of the many produced in the 1980s, purported to show that the DST was a
valid marker for endogenous depression by describing all the things that
nonsuppressors were not. In a 1986 study of 187 unipolar depressed inpatients
by Zimmerman and colleagues,12 nonsuppressors were shown to be
older, not as personality disordered, not as socially or maritally compromised,
and not as assailed with recent stressful events as suppressors. Moreover,
nonsuppressors had made fewer nonserious suicide attempts and were less likely
to have alcoholic or antisocial first-degree relatives.12 This
demonstrated that not only was melancholia innate to the organism, but that its
sufferers also experienced to a lesser degree all the comorbidities afflicting
reactive depressives and presumably driving their nonendogenous disorder.
However,
despite the excitement over the DST, which led to thousands of subjects being
studied and countless patients undergoing the test in conjunction with their
psychiatric treatment, there were harsh critics of the descriptive-biological
approach. In a scathing editorial in Biological
Psychiatry, Ros13 lambasted the theory
behind the test for its “reductionism.” “The depressed patient emits various
signals indicating his depressed state,” he wrote. “Some of these are
physiological, but not all. The assumption that the DST, which is a highly
transduced signal, is more real than the clinical signals is nonscientific and
ideologically based.”13
Ros’ skepticism proved
prophetic. Patients who looked melancholic frequently suppressed and
neurotic-appearing patients frequently did not suppress, although both types
had symptoms meeting criteria for major depressive disorder (MDD). In 1985,
Arana and colleagues1 showed that while the DST had a sensitivity of
78% in psychotic depressives, sensitivity dropped to only 44% if psychosis was
not present. Two years later, the American Psychiatric Association14
repudiated the DST on the basis of its lacking both sensitivity and specificity
as a diagnostic test for MDD. Like insulin-shock therapy and prefrontal
leukotomy, the DST seemed destined to become a historical anomaly.
The DST and
Risk of Suicide
Its lack of validity as a
diagnostic test notwithstanding, something significant appears to be happening
in the stress axis of depressed patients, which has continued to attract
research attention. While specific details still remain vague, the interplay
between the HPA axis and prefrontal serotonin systems—components of larger
brain modulatory networks—have come to be understood to modulate mood and
levels of impulsivity. The DST, therefore, has shown new promise in a different
psychiatric arena: identifying subjects at elevated suicide risk.
Coryell
and Schlesser,15 for example, reported findings on the mortality in
the ensuing years of 78 affectively ill subjects they had originally studied
between 1979 and 1981. Thirty-two had been DST nonsuppressors and 46 had been
suppressors. During an average of 10 years of follow-up, 8 of the 78 had
committed suicide. Seven of these eight were nonsuppressors, for a
nonsuppressor suicide prevalence of 27%, an order of magnitude greater than the
prevalence of 3% in the suppressor group.15 With a sample of 114
depressed patients almost evenly split between suppressor and nonsuppressors,
Bostwick and Warzecha16 replicated these findings. Of the seven
suicides in their sample during an average 14-year follow-up, all but one
occurred in nonsuppressors.
Clues to understanding these
findings are found in the functioning of the HPA axis in health and disease.
Under normal conditions, the HPA axis serves to maintain organism homeostasis.
The PVN of the hypothalamus releases corticotropin-releasing factor (CRF),
which stimulates the anterior pituitary to secrete adrenocorticotropic hormone
(ACTH), which in turn causes the adrenal cortex to release cortisol. Through a
negative feedback loop, the cortisol binds to hypothalamic receptors in a
two-level recognition system.10
The
first level involves mineralocorticoid receptors, high-potency receptors that
respond to low concentrations of cortisol to maintain optimal cellular
functioning in immune, metabolic, and autonomic nervous systems, among others.
This exquisitely tuned regulatory mechanism assures appropriate basal cortisol
levels through a typical day’s circadian fluctuations. When cortisol levels are
high, as occurs during acute physical or psychological threat perceived by the
cortex and funneled through the hypothalamus, the latter emits a burst of CRF.
The resultant cortisol outpouring from the adrenals saturates mineralocorticoid
receptors, and second-level glucocorticoid receptors are activated. Ineffective
and quiescent when cortisol is at baseline levels, these low-potency receptors,
when engaged in a cortisol-rich environment, have the capacity to trump the
organism’s homeostatic processes in favor of mobilizing energy reserves for the
fight-or-flight response. Heightened alertness and increased fuel supply to
muscles occur at the temporary expense of sleep, appetite, and libido. When
cortical input to the hypothalamus indicates the threat has passed,
glucocorticoid receptor-mediated negative feedback brings the HPA axis back to
baseline function.17
Both
basal regulatory activities of mineralocorticoid receptors and stress-management
functions of glucocorticoid receptors are essential to negotiating internal
requirements and external threats. When glucocorticoid receptor hyperactivity
becomes chronic, however, as it does in such conditions as Cushing’s syndrome
and severe depression, continuous high levels of cortisol exert a potent
influence on hormone and neurotransmitter receptor function, causing dramatic
affective and cognitive changes. Those familiar with behavioral side effects
prominent during exogenous high-dose corticosteroid administration for
multifarious medical and surgical conditions will appreciate similar findings
in the context of endogenous HPA axis disruption. These may include mood
extremes ranging from euphoria to depression, anxious and obsessional behavior,
insomnia, irritable and labile mood states, and cognitive disturbances in
perception, attention, concentration, and memory. All of these features can
complicate depressive states that result from either endogenous or iatrogenic
elevated corticosteroid levels. At their most extreme, these signs and symptoms
constitute the psychosis of certain depressive conditions. Indeed, while the
DST proved inadequately sensitive for general depressive states, it is more
reliably positive in psychotic depression,1 a fact which makes
intuitive sense given the potential role of elevated corticosteroids in
producing conditions in which agitation and psychosis may emerge. As early as
1982, Reus18 observed the clinical distinctiveness of patients with
hypercortisolemia:
Regardless of the diagnosis appended, the failure to suppress cortisol
following the DST appears to identify subjects who are more symptomatic both
objectively and subjectively at admission. The most significant differences
were an increased incidence of sleep disorder, feelings of anxiety, and
thoughts of suicide.
Chronically
elevated corticosteroids can result in permanent brain changes,18,19
a fact that has important implications for stress responsiveness up to and
including susceptibility to suicidal states. Such hypercortisolemic baselines
can have developmental roots. Studies in neonatal rats removed from their
mothers; monkeys raised by stressed, neglectful mothers; and mammals raised
under stressful conditions have demonstrated HPA axis overdrive, as manifested
in increased CRF receptors throughout the axis, increased
corticotropin-releasing hormone, and ACTH levels with stress, increased
cortisol at baseline, increased susceptibility to stress, and alterations in
brain function.20-24
Brain
damage in the form of corticosteroid-induced hippocampal atrophy yields reduced
inhibitory capacity characterized by a baseline trait of mild hyperarousal.
Under stress, this hyperarousal balloons into more severe arousal states than
found in subjects with normal hippocampal volume.25 This implies
that abuse or neglect may produce permanent changes in the developing brain,
according to Nemeroff.25 These changes chronically boost the input
of and responsiveness to CRF, and therefore increase the lifetime vulnerabilty
of subjects to depression.25 Certainly, histories of childhood abuse
and failures in nurturance figure prominently in clinical histories of patients
with dysthymia or many personality disorders. In patients with chaotic or
neglectful developmental histories, genetic predispositions to depression, or
both, superimposed acute stress can throw them into depressed or suicidal
states.26,27
A further correlate of the
HPA axis gone awry is a reduction in ventral prefrontal cortex (PFC) serotonin
levels when cortisol levels are high.28 Associated with damping
impulsive and angry behavior, as well as regulating mood, the ventral PFC has
been the brain region most closely linked to the monoamine hypothesis of
depression in vogue since the 1960s, even as this theory grows increasingly
simplistic as knowledge of neural networks and the complexity of reciprocal
neurotransmitter interactions swells.
In 1994, Dinan29
proposed that reduced ventral PFC serotonin was actually a downstream effect of
HPA axis overdrive. Mann,28 a prominent investigator of ventral PFC
activity in suicidal states, also proposed a “stress-diathesis” model of
suicide in which developmental failures and genetic predispositioning
correlates of HPA axis abnormalities are the substrates against which a
stressor invokes the low-serotonin state driving impulsive acts against others
or the self.28
While the exact mechanisms
by which the HPA axis modulates ventral PFC activity have not yet been
elucidated, additional correlations continue to emerge. Van Praag,30 for example, noted how prefrontal regions are critical in damping acute stress
reactors via negative feedback, how low serotonin in these regions is linked to
disturbed anxiety and aggression regulation, and how stress—presumably HPA-axis
induced—can trigger increased anxiety extending all the way up to the
self-directed aggression of suicidal behavior. Westrin and Nimeus31
also showed that nonsuppressors with low levels of cerebrospinal fluid 5-HIAA,
a serotonin metabolite, had higher scores on the Suicide Assessment Scale than
either nonsuppressors with normal 5-HIAA or suppressors with either low or
normal 5-HIAA.
Conclusion
The DST
may prove to be a neuroendocrine window to suicidal propensity. Studies by
Coryell and Schlesser15 and Bostwick and Warzecha16 support HPA axis dysregulation not as a psychiatric diagnostic tool but rather
as an indicator of propensity toward lethal behavior. While DST nonsuppressor
status will not pinpoint specific patients who will kill themselves and does
not yet have clinical relevance in primary care settings, it does hold the
promise of identifying a subgroup of patients at greatly elevated risk. Studies
with larger numbers of subjects are needed to confirm the findings of these two
small efforts. When depressive episodes in such patients assume the agitated or
psychotic proportions suggestive of HPA-axis overdrive, they should be closely
monitored for suicidality. In medical inpatients whose treatment includes
high-dose corticosteroids, the emergence of affective or behavioral instability
should be taken seriously, since impulsivity and disinhibition are closely
linked to completed suicide while in the hospital. Psychiatric and
nonpsychiatric providers alike should respond to anxiety or agitation,
particularly in patients with this HPA axis abnormality, to avert suicidal
crises. PP
References
1. Arana GW, Baldessarini RJ, Ornsteen M. The dexamethasone suppression test for diagnosis and prognosis in psychiatry. Commentary and review. Arch Gen Psychiatry . 1985;42(12):1193-1204.
2. Kino T, Chrousos GP. Glucocorticoid and mineralocorticoid resistance/hypersensitivity syndromes. J Endocrinol . 2001;169(3):437-445.
3. Cannon WB. The Wisdom of the Body . New York, NY: W.W. Norton; 1939.
4. Cushing H. Psychic disturbance associated with disorders of the ductless glands. Am J Insanity. 1913;69(5):965-990.
5. Varghese FP, Brown ES. The Hypothalamic-pituitary-adrenal axis in major depressive disorder: a brief primer for primary care physicians. Prim Care Companion J Clin Psychiatry . 2001;3(4):151-155.
6. Sachar EJ. Corticosteroids in depressive illness. II. A longitudinal psychoendocrine study. Arch Gen Psychiatry . 1967;17(5):554-567.
7. Sachar EJ, Hellman L, Fukushima DK, Gallagher TF. Cortisol production in depressive illness. A clinical and biochemical clarification. Arch Gen Psychiatry . 1970;23(4):289-298.
8. Bunney WE, Mason JW, Roatch JF, Hamburg DA. A psychoendocrine study of severe psychotic depressive crises. Am J Psychiatry . 1965;122(1):72-80.
9. Gibbons JL. The secretion rate of corticosterone in depressive illness. J Psychosom Res . 1967;10:263-266.
10. Butler PW, Besser GM. Pituitary-adrenal function in severe depressive illness. Lancet . 1968;1(7554):1234-1236.
11. Carroll BJ, Feinberg M, Greden JF, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry . 1981;38(1):15-22.
12. Zimmerman M, Coryell W, Pfohl B. The validity of the dexamethasone suppression test as a marker for endogenous depression. Arch Gen Psychiatry . 1986;43(4):347-355.
13. Ros CA. Biological tests for mental illness: their use and misuse. Biol Psychiatry . 1986;21(5-6):431-435.
14. The APA Task Force on Laboratory Tests in Psychiatry. The dexamethasone suppression test: an overview of its current status in psychiatry. Am J Psychiatry . 1987;144(10):1253-1262.
15. Coryell W, Schlesser M. The dexamethasone suppression test and suicide prediction. Am J Psychiatry . 2001;158(5):748-753.
16. Bostwick JM, Warzecha J. The dexamethasone suppression test as a predictor of suicide risk. Mayo Clin Proc . In press.
17. Mackin P, Young AH. The role of cortisol and depression: exploring new opportunities for treatment. Psychiatric Times . 2004;21(6):92-95.
18. Reus VI. Pituitary-adrenal disinhibition as the independent variable in the assessment of behavioral symptoms. Biol Psychiatry . 1982;17(3):317-326.
19. Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab . 2002;13(9):373-380.
20. Vythilingam M, Heim C, Newport J, et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry . 2002;159(12):2072-2080.
21. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology . 2000;23(5):477-501.
22. Essex MJ, Klein MH, Cho E, Kalin NH. Maternal stress beginning in infancy may sensitize children to later stress exposure: effects on cortisol and behavior. Biol Psychiatry . 2002;52(8):776-784.
23. Kaufman J, Plotsky PM, Nemeroff CB, Charney DS. Effects of early adverse experiences on brain structure and function: clinical implications. Biol Psychiatry . 2000;48(8):778-790.
24. Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci . 2001;24:1161-1192.
25. Nemeroff CB. The neurobiology of depression. Sci Am . 1998;278(6):42-49.
26. Newport DJ, Stowe ZN, Nemeroff CB. Parental depression: animal models of an adverse life event. Am J Psychiatry . 2002;159(8):1265-1283.
27. Arato M, Banki CM, Nemeroff CB, Bissette G. Hypothalamic-pituitary-adrenal axis and suicide. Ann N Y Acad Sci . 1986;487:263-270.
28. Mann JJ. The neurobiology of suicide. Nat Med . 1998;4(1):25-30.
29. Dinan TG. Glucocorticoids and the genesis of depressive illness. A psychobiological model. Br J Psychiatry . 1994;164(3):365-371.
30. van Praag HM. Can stress cause depression? Prog Neuropsychopharmacol Biol Psychiatry . 2004;28(5):891-907.
31. Westrin A, Nimeus A. The dexamethasone suppression test and CSF-5-HIAA in relation to suicidality and depression in suicide attempters. Eur Psychiatry . 2003;18(4):166-171.